
La EB es muy heterogénea clínicamente y varían mucho los grados de severidad. Se han descrito mutaciones pateogénicas (que causan enfermedad) en más de 20 genes asociados a la EB. Muchos de estos genes se traducen en proteínas como colágenos, lamininas e integrinas. Estas proteína presentes en la piel, aseguran su integridad y resistencia y dependiendo de qué afectación presente cada proteína (ausente, reducida, no funcional, etc.), y de su localización en las distintas capas de la piel, la severidad y manifestación de la enfermedad varía.
A modo introductorio, la EB se clasifica en 4 tipos: Simple, Juntural, Distrófica y Kindler. En cada tipo se han descrito una o más proteínas afectadas. En la siguiente imagen se muestran en qué parte de la piel se encuentran las distintas proteínas afectadas en los distintos tipos de Epidermólisis bullosa.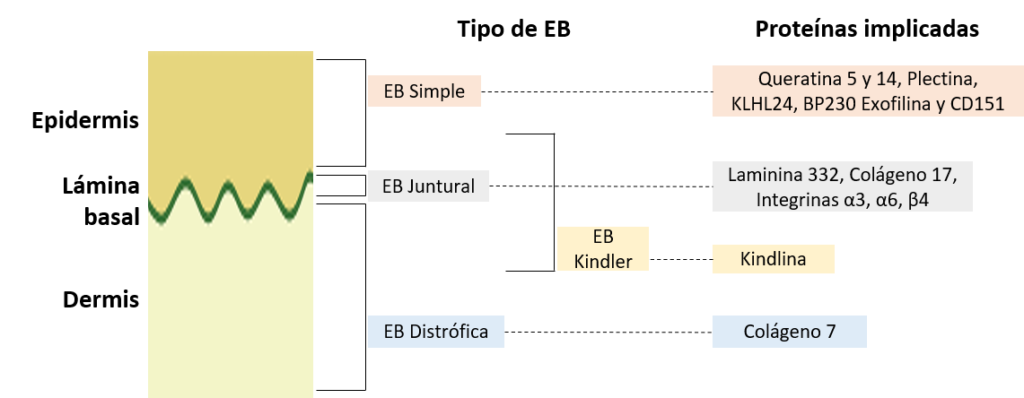
La (re-)clasificación de la enfermedad se lleva a cabo cada ciertos años, cuando se han identificado nuevos genes y subtipos clínicos. La clasificación de los 4 tipos clásicos y los subtipos actuales, se basan en el artículo publicado en Febrero del 2020, Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility (Has C., et al) por un comité experto en EB.
Con más de 30 subtipos, queda claro que la EB es clínicamente heterogénea incluyendo un amplio abanico de severidad. Para determinar el tipo y subtipo de EB en recién nacidos o personas con síntomas más leves, se necesita hacer pruebas diagnósticas en el laboratorio. En los artículos siguientes explicaremos los signos y síntomas de los tipos más comunes.